Neurotrasmissione
La neurotrasmissione è l’insieme di tappe che ogni neurotrasmettitore segue dalla sua sintesi fino all’effetto finale; esse sono:
- Sintesi.
- Rilascio.
- Interazione con i recettori e avvio dell’attività postsinaptica.
- Catabolismo e/o dissipazione.
Sintesi del neurotrasmettitore I neurotrasmettitori non peptidici (piccole molecole) vengono sintetizzati principalmente nella regione delle terminazioni assoniche e accumulati all’interno delle vescicole sinaptiche.
I neurotrasmettitori peptidici (o i precursori peptidici) si trovano in grandi vescicole dense che vengono trasportate lungo l’assone dal loro luogo di sintesi nel corpo cellulare.
Rilascio del neurotrasmettitore Il potenziale d’azione determina la liberazione sincrona di parecchie centinaia di quanti di neurotrasmettitore. Questo processo viene iniziato dalla depolarizzazione della terminazione assonica; una tappa critica per molte terminazioni nervose, ma non per tutte, è l’influsso di Ca2+, che entra nel citoplasma assonico e consente la fusione tra la membrana assoplasmatica e quelle vescicole situate in stretta prossimità della membrana stessa. Il contenuto delle vescicole, compresi enzimi e altre proteine, viene quindi riversato all’esterno attraverso un processo di esocitosi. Le vescicole sinaptiche possono subire un processo di esocitosi completa, con totale fusione e conseguente endocitosi, oppure possono formare un poro transitorio che si chiude subito dopo il passaggio del neurotrasmettitore.
Le vescicole sinaptiche sono localizzate in determinate aree sottostanti la membrana plasmatica presinaptica, denominate zone attive; queste sono spesso allineate con gli apici delle pieghe postsinaptiche.
La sinaptobrevina (VAMP) si associa con la proteina di membrana SNAP-25 e la sintaxina 1 per formare un complesso che inizia o guida il processo di fusione della vescicola con la membrana plasmatica.
L’esocitosi istantaneamente scatenata dal Ca++ sembra essere mediata da una diversa famiglia di proteine, le sinaptotagmine.
Interazione del neurotrasmettitore con i recettori einizio dell’attività postsinaptica I trasmettitori diffondono attraverso lo spazio intersinaptico e interagiscono con recettori specializzati sulla membrana postsinaptica; questo evento spesso risulta in un aumento localizzato della permeabilità ionica (o conduttanza) della membrana. I cambiamenti di permeabilità possono essere di tre tipi:
- Un aumento generalizzato della conduttanza ai cationi (in particolare Na+, ma a volte Ca++), che risulta in una depolarizzazione localizzata della membrana, cioè un potenziale postsinaptico eccitatorio (EPSP).
- Un aumento selettivo della permeabilità agli anioni, generalmente Cl–, che risulta in una stabilizzazione o meglio in una iperpolarizzazione della membrana che costituisce un potenziale postsinaptico inibitorio (IPSP).
- Un’aumentata permeabilità al K+. Poiché il gradiente del K+ è diretto verso l’esterno della cellula, ne consegue una iperpolarizzazione e stabilizzazione della membrana (un IPSP).
Se un EPSP eccede un determinato livello soglia, è in grado di iniziare e propagare un potenziale d’azione in un neurone postsinaptico o un potenziale d’azione muscolare in un muscolo scheletrico o nel muscolo cardiaco, attivando i canali voltaggio-sensibili nelle immediate vicinanze.
In alcuni tipi di muscolatura liscia, nella quale la propagazione dell’impulso è minima, un EPSP può provocare un aumento nella frequenza di depolarizzazione spontanea, indurre il rilascio di Ca2+, e aumentare il tono muscolare; nelle cellule ghiandolari, l’EPSP dà inizio alla secrezione attraverso la mobilizzazione del Ca++.
Un IPSP, che è osservabile nei neuroni e nella muscolatura liscia, ma non nella muscolatura scheletrica, tenderà a opporsi a potenziali d’azione eccitatori iniziati contemporaneamente da altre fonti neuronali nello stesso sito. La sommatoria di questi potenziali determina la natura della risposta che ne deriva.
È possibile che un neurotrasmettitore liberato dai neuroni presinaptici agisca su recettori presinaptici che possono essere distinti in eterorecettori e autorecettori.
- Gli eterorecettori modulano (in senso positivo o negativo) il rilascio di un altro neurotrasmettitore; ad esempio, l’acetilcolina può legarsi ai recettori M2 o M4 delle fibre adrenergiche inibendo il rilascio di noradrenalina.
- Gli autorecettori inibiscono il rilascio dello stesso neurotrasmettitore che li ha attivati; ad esempio, l’acetilcolina può legarsi ai recettori M2 o M4 presinaptici inibendo il suo stesso rilascio.
I recettori presinaptici presenti sul terminale colinergico possono essere:
- Autorecettori, la cui attività è regolata dallo stesso neurotrasmettitore (l’acetilcolina, ACh) e si suddividono in:
- Nicotinico (così detto perché attivato dalla nicotina), che attivato favorisce il rilascio di ACh (feedback positivo, produce una depolarizzazione del terminale importante per la liberazione del neurotrasmettitore).
- Muscarinico (così detto perché attivato dalla muscarina), che attivato inibisce il rilascio di ACh (feedback negativo, dato che il recettore M2 è accoppiato a proteine G di tipo inibitorio che riducono la concentrazione intracellulare di cAMP ed iperpolarizzano la membrana attraverso l’attivazione dei canali al potassio attivate dalla subunità βgdella proteina G ed iperpolarizzano il terminale riducendo il livello di fosforilazione intracelulare dato che viene a mancare l’attivazione di protein-chinasi da parte del cAMP data la sua concentrazione ridotta nel terminale).
- Eterorecettori, non attivati dal neurotrasmettitore che si libera nella sinapsi ma da altri mediatori endogeni come la dopamina, le encefaline, la serotonina che modulano il rilascio del neurotrasmettitore con un meccanismo a feedback positivo o negativo a secondo del tipo di recettore coinvolto. Questi recettori possono essere di tipo inibitorio o stimolatorio.
- Di tipo inibitorio (modulazione a feedback negativo):
- Recettori di tipo µ, per gli oppioidi, attivati da sostanze che si ricavano dall’oppio come la morfina e l’eroina, un derivato acetilato della morfina, la tebaina ed altri, ed esistono anche dei ligandi endogeni per questi recettori rappresentati dalle encefaline. L’attivazione di recettori di tipo µ inibisce il rilascio di ACh utilizzando un meccanismo di trasduzione molto simile ai recettori muscarinici M2 che riducono il livello di fosforilazione dei terminali ed iperpolarizzano la membrana opponendosi alla liberazione del neurotrasmettitore. Tali recettori sono bersaglio di farmaci utilizzati a livello periferico per la loro attività costipante attraverso l’inibizione del rilascio di ACh nei gangli intramurali intestinali, cioè rallentando la motilità intestinale (quindi sono usati come antidiarroici). Un tale composto è il difenossilato.
- Recettori di tipo adrenergico, sono recettori α2 non importanti per farmaci usati allo scopo terapeutico di inibire l’ACh, ma la cui stimolazione può causare effetti indesiderati come la stipsi dovuta alla sua azione sui recettori α2 adrenergici a livello del plesso mienterico intestinale di Auerbach.
- Recettori dopaminergici, che appartengono ai recettori D2. Gli antagonisti del recettore dopaminergico sono farmaci procinetici e favoriscono il rilascio di ACh. Un antagonista del recettore dopaminergico che agisce a livello sistemico originerà l’effetto opposto dato che viene antagonizzato l’effetto mediato dalla dopamina e sono quei procinetici usati nei disturbi della motilità gastrointestinale.
- Recettori per la serotonina (5HT1 e 5HT2) e recettori per il GABAA.
- Di tipo stimolatorio:
- Recettori di tipo D1 che sono usati in terapia ed hanno una regolazione positiva e favoriscono il rilascio di ACh.
- Recettori per la serotonina, bersaglio di farmaci utilizzati come procinetici. La serotonina media i suoi effetti attraverso recettori serotoninergici tra cui ce ne sono di 5 sottotipi; a questo livello il recettore 5HT4, quando attivato, favorisce il rilascio di ACh. Ne consegue che farmaci che si comportano come agonisti di questo recettore, favoriscono il rilascio di ACh e sono utilizzati come farmaci procinetici.
- Recettore per la colecistochinina (CCK).
- Di tipo inibitorio (modulazione a feedback negativo):
Catabolismo e/o dissipazione del neurotrasmettitore A livello delle sinapsi sono presenti meccanismi che determinano il catabolismo, quindi la degradazione, o la dissipazione (o entrambi) del neurotrasmettitore.
La degradazione avviene mediante specifici enzimi in grado di degradare il neurotrasmettitore (ad esempio, l’acetilcolinesterasi per la degradazione dell’acetilcolina).
La dissipazione può avvenire tramite due processi: diffusione del neurotrasmettitore o sua ricaptazione (o reuptake) da parte dei terminali assonali (ad esempio, una parte delle catecolamine è allontanata dalla sinapsi in questo modo).
Articolo creato l’11 aprile 2011.
Ultimo aggiornamento: vedi sotto il titolo.
Potrebbe anche piacerti:

Examen oficial MIR 2008
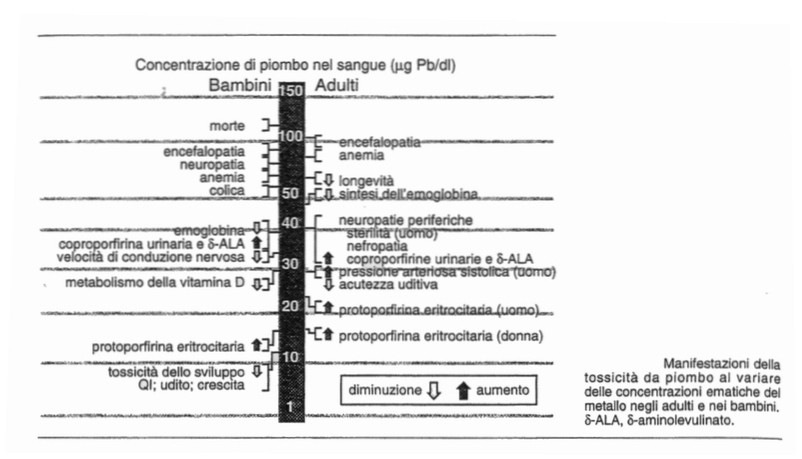
Avvelenamento da piombo